
A novel link between vitamin D deficiency, beta-amyloid protein signaling, and aging in the development of parathyroid tumors
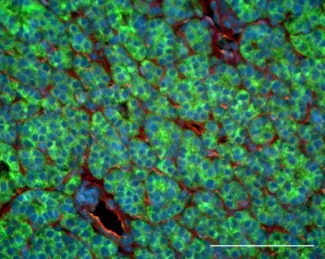
The amyloid precursor protein APP (stained in green) is highly expressed in many parathyroid tumors.
Vitamin D deficiency is a widespread problem among the elderly and can precipitate sequelae that are particularly harmful to this vulnerable population. Hyperparathyroidism (HPT) is commonly associated with low vitamin D status, and the disruption in calcium homeostasis that is the cardinal feature of this disease can lead to a range of deleterious outcomes among the elderly, including loss of bone density, muscle weakness, and increased risk of falls and pathological fracture. As the primary endocrine organs responsible for hormonal control of serum calcium levels, the parathyroid glands are a known target for the actions of vitamin D, yet there are major gaps in our understanding of how vitamin D deficiency and attenuated vitamin D receptor (VDR) signaling contribute to the etiology and clinical presentation of HPT. Several key findings from our group suggest a testable new model for vitamin D-mediated actions in the parathyroid gland. First, we showed that the GABA B1 receptor (GABAB1R) can form heterodimeric complexes with CaSR, promoting parathyroid hormone (PTH) hypersecretion by opposing the coupling of CaSR with its obligate downstream G-protein effectors Gq/11 and Gi. Second, building upon a recent report demonstrating that soluble peptide derivatives of the amyloid precursor protein (APP) can bind and activate GABAB1R in neurons, we found that the APP-derived peptide Ab1-42 can increase maximal PTH secretion by parathyroid tissue in a CASR- and GABAB1R-dependent manner. VDR expression and serum vitamin D levels are inversely correlated with the relative abundance of APP, Ab1-42, the g- and b-secretases required for Ab1-42 production, and the phosphorylated form of the microtubule associated protein Tau (pTau). Functionally, ablation of APP in the parathyroid abrogates the development of HPT in VDR KO mice, and inhibitors of Tau phosphorylation can block the ability of Ab1-42 to promote PTH hypersecretion. These data suggest that HPT driven by loss of VDR activity could arise at least in part through unregulated expression of Ab1-42 and pTau. Based on these findings, we hypothesize that aging-induced increases in Ab1-42-mediated signaling drive tonic PTH hypersecretion and that vitamin D deficiency exacerbates HPT disease severity by relieving suppression of Ab1-42 production and Tau/pTau expression. We are currently testing this model through three complementary experimental approaches: (1) delineating the molecular actions of Ab1-42 on CaSR, GABAB1R, and downstream signaling events that promote PTH hypersecretion; (2) determinng whether blocking the production or activity of Ab1-42 and Tau delays HPT development in a murine model of CaSR insufficiency; and (3) elucidating the causal relationship between VDR and Ab1-42/pTau signaling in the parathyroid. By defining the contributions of the CaSR/GABAB1R/Ab1-42 signaling axis to PTH hypersecretion, this work will provide a clearer understanding of the unexpected connection between b-amyloid peptides, vitamin D status, and parathyroid gland function.
This work is supported by NIH grants 1R21AG070721 and 1RF1AG075742.
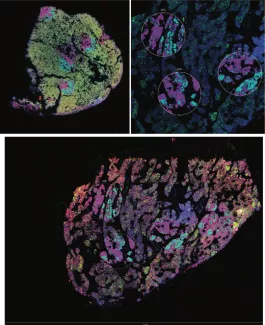
Digital spatial profiling of human parathyroid tissue
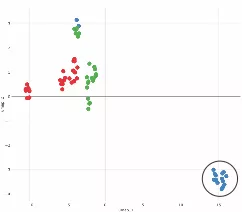
Parathyroid tumors from patients with vitamin D deficiency (blue symbols, circled) are molecularly distinct from normal tissue (green symbols) and from PHPT patients who have normal vitamin D levels (red symbols).
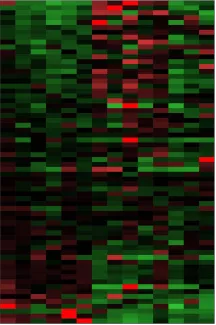
A heat map of gene expression levels helps visualize patterns shared by tumors compared to normal tissues.